

| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website http://www.wjnu.org |
Case Report
Volume 1, Number 4-5, October 2012, pages 127-129
Two Male Siblings With Alport Syndrome
Anggoro B. Hartopoa, b, d, Iri Kuswadib, Hartonoc
aDepartment of Cardiology and Vascular Medicine, Faculty of Medicine Gadjah Mada University, Dr. Sardjito Hospital, Yogyakarta, Indonesia
bDepartment of Internal Medicine, Faculty of Medicine Gadjah Mada University, Dr. Sardjito Hospital, Yogyakarta, Indonesia
cDepartment of Ophthalmology, Faculty of Medicine Gadjah Mada University, Dr. Sardjito Hospital, Yogyakarta, Indonesia
dCorresponding author: Anggoro B. Hartopo, Department of Cardiology and Vascular Medicine, Faculty of Medicine Gadjah Mada University, Dr. Sardjito Hospital, Jalan Kesehatan no. 1, Sekip, Yogyakarta, 55284, Indonesia
Manuscript accepted for publication October 2, 2012
Short title: Alport Syndrome
doi: https://doi.org/10.4021/wjnu43w
| Abstract | ▴Top |
Alport syndrome is inherited disease affected young population and culminates in end-stage kidney disease. X-linked Alport syndrome causes end-stage kidney disease in young male patients, sometime without prominent clinical manifestation beforehand due to its heterogeneous phenotype. We report two male siblings with end-stage kidney disease supposedly caused by Alport syndrome, without previous reported signs and symptoms.
Keywords: Kidney; Alport syndrome; Inherited disease
| Introduction | ▴Top |
Hereditary nephritis or Alport syndrome is an inherited disease characterized clinically by progressive kidney disease accompanied with sensorineural hearing loss and ocular abnormalities [1]. Alport syndrome accounts for 0.3 to 2.3% of end-stage kidney disease [2]. It is related to the mutations in type IV collagen genes [3]. Type IV collagen chains form network which presents in glomerular basement membrane, ear, eye, and lung [3].
Gen frequency of Alport syndrome is estimated from 1:5,000 to 1:10,000 [4]. Around 85% of Alport syndrome is X-linked disease, which affects male patients in severe form [2]. In kidney, proteinuria first appears in childhood and progress to end-stage kidney disease in early adulthood [2].
Here we present two cases of end-stage kidney diseases affected two young male siblings which suggestively caused by hereditary nephritis or Alport syndrome. The patients did not reported any signs and symptoms before the diagnose of end-stage kidney disease.
| Case Report | ▴Top |
We report cases of two male siblings who suffered from end-stage kidney disease. The first patient was 25-year-old male. He presented in internal medicine outpatient clinic of our hospital with chief complaint of nausea and intense fatigue. He felt this symptoms since 4 months ago, accompanied by abdominal discomfort and reduced micturition. Physical examination showed alert general condition, blood pressure 140/90 mmHg, regular pulse 100 ×/min, pale conjunctiva, unremarkable cardiac examination and no ankle oedema. We ordered a laboratory examination and found the result: haemoglobin 5.4 g/dL, creatinine 30.16 mg/dL, blood urea nitrogen 195.4 mg/dL and potassium 4.66 mEq/L. Calculated clearance creatinin test based on Cockroft-Gault formula was 2.9. Blood gas analysis revealed pH 7.181 and HCO3- 5.0. Being alerted of the presence of renal disease, we ordered kidney ultrasound examination which showed symmetrically reduced kidneys (Fig. 1), radiogram BNO examination which showed the absence of radioopaque stones in urinary tracts and renogram examination which concluded non-functionality of both kidneys. We diagnosed the patients with stage V chronic kidney disease with current problem of severe anaemia, metabolic acidosis and uremic syndrome. We performed urgent haemodialysis with packed-red cell transfusion and treated the patients with haemodialysis with regular basis.
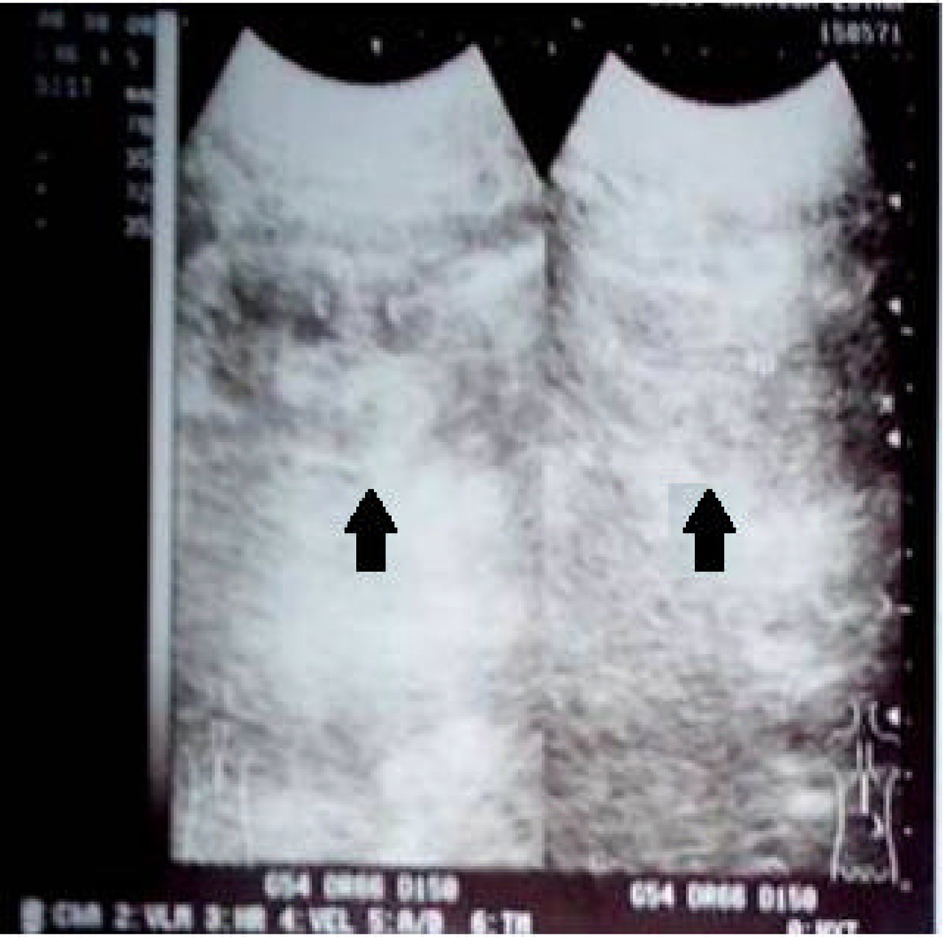 Click for large image | Figure 1. Kidney ultrasound showed symmetrically reduced kidneys (arrow). |
The second patient was 27-year-old male, who was the older brother of the first patient. He had been undergoing routine haemodyalisis in our haemodyalisis unit since 2 years ago, at age of 25. He recalled the first symptoms similar to that of his younger brother, nausea and fatigue. Laboratory examination at that time showed haemoglobin 5.7 g/dL, creatinine 26.68 mg/dL, blood urea nitrogen 147.4 mg/dL and potassium 3.5 mEq/L. Calculated clearance creatinin test based on Cockroft-Gault formula was 3.3. Kidney ultrasound revealed symmetrically reduced kidneys. Radiogram BNO examination showed nonexistence radioopaque stones in urinary tracts. Renogram examination revealed non-functionality of both kidneys. He had been diagnosed with stage V chronic kidney disease since then and until current time had been treated with continuous ambulatory peritoneal dyalisis (CAPD).
We pursued the hereditary nephritis or Alport syndrome as an underlying cause of nephropathy in our patients. To this end, we sought ophthalmologist and ENT specialist consultations. For the first patients, ophthalmologist examination showed refraction disorder myopia, without signs of anterior lenticonus or perimacular flecks. ENT examination showed sensorineural hearing loss in both right and left ear in audiometric test. For the second patient, ophthalmologist examination also showed refraction disorder myopia. ENT examination was not performed for this patient. Skin or kidney biopsy was not conducted, because the patients refused the procedure.
Family history revealed that the patients’ mother and father had third cousin marriage. They had four children, three males and one female. The oldest and second male children currently suffered from end-stage kidney disease. Patients’ uncle, from mother side, had undergone hemodyalisis and recently passed away. The patients have two other siblings, one younger brother and one sister. The younger brother, age 20, was in healthy condition. We screened him for possibility of hereditary nephritis. Clinical and laboratory examination showed no abnormality. Urinalysis examination showed occult haematuria (positive for blood urine and 3 - 8 erythrocytes/high-power field (2 - 3 dismorphic erythrocytes). The sister, age 23, was also in healthy condition. We also screened her for possibility of hereditary nephritis. Clinical and laboratory examination showed no abnormality. Urinalysis examination showed occult haematuria (positive for blood urine and 3 - 5 erythrocytes/high-power field (0 - 2 dismorphic erythrocytes)). We also checked the mother and revealed no abnormality observed. We encouraged them to reexamine in the next 6 month.
| Discussion | ▴Top |
Alport syndrome is a primary glomerular basement membrane disease occurring from type IV collagen genes mutation [3]. Six different genes encode six distinctive type IV collagen chain proteins [5]. These genes are located in three chromosomes: COL4A1 and COL4A2 genes at chromosome 13, COL4A3 and COL4A4 genes at chromosome 2 and COL4A5 and COL4A6 genes on chromosome X [5]. The mutations of these genes caused disruption of type IV collagen synthesis and network formation [5]. Mutation in the COL4A5 gene which encodes collagen α-5(IV) chain cause X-linked Alport syndrome, which mode of inheritance accounts for 85% of all Alport syndromes [2].
X-linked Alport syndrome is clinically heterogeneous [6]. The natural history of the kidney disease manifestation varies. Age at development of end-stage kidney disease differs and in males ranges between second and third decade of life [7]. In milder involvement, end-stage kidney disease may be postponed until the fifth or sixth decade [7]. Both our patients developed end-stage renal disease at age 25, which indicated severe phenotype involvement. Unfortunately without any previous signs and symptoms that prompted our patients sought medical treatment. They visited our hospital and diagnosed with end-stage kidney disease which needed kidney replacement therapy.
In addition to kidney involvement, sensorineural deafness and ocular abnormality, namely anterior lenticonus, cataract, and maculopathy, have been reported in X-linked Alport syndrome [7]. These extrarenal involvements also vary in commencement and frequency [7]. Our investigation of the patients revealed sensorineural hearing loss and refraction disorder, myopia. Myopia may be a manifestation of Alport syndrome due to anterior lenticonus [8]. Clinical manifestation and family history of our patients suggested of Alport syndrome [4]. However, we neither performed histopathology examination because of patients refusal nor genetic diagnosis because its complexity and unavailability in our hospital.
| References | ▴Top |
- Grunfeld JP. The clinical spectrum of hereditary nephritis. Kidney Int. 1985;27(1):83-92.
doi pubmed - Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009;20(6):1210-1215.
doi pubmed - Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003;348(25):2543-2556.
doi pubmed - Pirson Y. Making the diagnosis of Alport's syndrome. Kidney Int. 1999;56(2):760-775.
doi pubmed - Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15(10):2514-2527.
doi pubmed - Kashtan CE. Alport syndromes: phenotypic heterogeneity of progressive hereditary nephritis. Pediatr Nephrol. 2000;14(6):502-512.
doi pubmed - Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, Schrier RW. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010;21(5):876-883.
doi pubmed - Gupta A, Ramesh Babu K, Srinivasan R, Mohanty D. Clear lens extraction in Alport syndrome with combined anterior and posterior lenticonus or ruptured anterior lens capsule. J Cataract Refract Surg. 2011;37(11):2075-2078.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Nephrology and Urology is published by Elmer Press Inc.