

| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website http://www.wjnu.org |
Case Report
Volume 3, Number 1, March 2014, pages 41-48
A Case of Collapsing Glomerulopathy Secondary to Romidepsin Therapy in a Patient With Subcutaneous Panniculitis-Like T-Cell Lymphoma
Sakshi Kapura, e, Ryan P. Brownb, Michael B. Stokesc, Miles B. Levind
aDepartment of Internal Medicine, Overlook Medical Center, 99 Beauvoir Ave, Summit, New Jersey 07902, USA
bDivision of Nephrology, Overlook Medical Center, 99 Beauvoir Ave, Summit, New Jersey 07902, USA
cDepartment of Pathology and Cell biology, Columbia University and Medical Center, 630 West 168th Street, New York, NY 10032, USA
dDivision of Pathology, Overlook Medical Center, 99 Beauvoir Ave, Summit, New Jersey 07902, USA
eCorresponding author: Sakshi Kapur, Department of Internal Medicine, Overlook Medical Center, 99 Beauvoir Ave, Summit, New Jersey 07902, USA
Manuscript accepted for publication December 20, 2013
Short title: Glomerulopathy Secondary to Romidepsin Therapy
doi: https://doi.org/10.14740/wjnu132w
| Abstract | ▴Top |
We report a case of a 44-year-old female with subcutaneous panniculitis-like T-cell lymphoma, who developed acute renal failure and nephrotic syndrome following treatment with romidepsin. Renal biopsy revealed collapsing variant of focal segmental glomerulosclerosis (FSGS), with good clinical response to high dose steroids. Although there is an association between paraneoplastic glomerulonephritides and lymphoma, in this case the temporal association and response to therapy, despite progression of the lymphoma, favor FSGS secondary to romidepsin therapy. To our knowledge, this is the first reported case of collapsing FSGS secondary to romidepsin therapy.
Keywords: Focal segmental glomerulosclerosis; Romidepsin; Paraneoplastic glomerulonephritides
| Introduction | ▴Top |
Romidepsin, a histone deacetylase inhibitor, is used to treat cutaneous T-cell lymphoma in patients who have failed at least one prior systemic therapy. Glomerular injury secondary to romidepsin has not been previously described. Our patient developed nephrotic syndrome collapsing variant focal segmental glomerulosclerosis (FSGS) immediately following treatment with romidepsin. Although her lymphoma continued to progress on chemotherapy, the nephrotic syndrome responded well to steroids. Various agents such as bisphosphonates, heroin, lithium, interferons and anthracyclines are known to cause collapsing FSGS. Our case highlights glomerular injury (collapsing FSGS) as a potential side effect of romidepsin therapy.
| Case Report | ▴Top |
A 44-year-old African American female presented with a one-week history of fever, chills and a mass in the right submandibular region. Six weeks previously, she had noticed two soft tissue masses in her left leg. She also reported a fifty pound weight loss and occasional night sweats over the last six months. She had an extensive family history of ovarian cancer and was tested positive for BRCA mutation.
Head and neck exam revealed a mass, measuring 3 х 2 cm, in right submandibular region. Two masses, one in left upper thigh and other behind left knee, measuring about 5 х 3 cm and 3 х 2 cm, respectively, were noted. These masses were tender on palpation, and had marked erythema and induration surrounding them. Both lymphadenopathy and hepato-splenomegaly were absent on exam.
Further evaluation revealed a hemoglobin of 10.7 g/dL, white blood cell count of 4.3 х 103/µL, and platelet count of 340 х 103/µL. Blood urea nitrogen, serum creatinine and estimated glomerular filtration rate were 11 mg/dL, 0.9 mg/dL and > 60 mL/min/1.73 m2, respectively. Urinalysis was within normal limits. Sedimentation rate and C-reactive protein were 101 mm/hr and 158 mg/L, respectively. Hepatitis profile and HIV 1 and 2 antibodies were non-reactive.
Excisional biopsy of the masses revealed findings compatible with subcutaneous panniculitis-like T-cell lymphoma, alpha/beta positive (Fig. 1). The corresponding flow cytometry revealed CD8+/CD56- phenotype, and chromosomal analysis of the tissue was unremarkable.
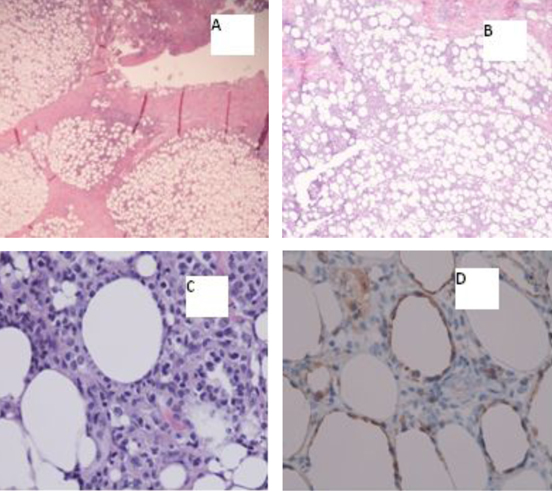 Click for large image | Figure 1. A) Inflammatory subcutaneous infiltrate with lobular pattern (20х); B) Inflammatory subcutaneous infiltrate with lobular pattern (40х); C) Atypical/neoplastic cells “rimming fat lobules” (400х); D) Cytotoxic immunohistochemical stain “perforin” labels the neoplastic cells, and again highlights the fat rimming (400х). |
Bone marrow biopsy results showed a mildly hypercellular marrow (70%) with trilineage hematopoiesis, normal erythroid maturation, M:E ratio of 4:1, normal megakaryocytes, scattered lymphocytes consisting mostly of CD3+ T-cells and CD4:CD8 ratio of 2:1. The aspirate was unimpressive for hemophagocytic syndrome. Both flow cytometry and chromosomal analysis of bone marrow aspirate were unremarkable.
Whole body PET-CT scan showed multiple areas of hypermetabolic activity in submandibular region, inferior aspect of chin, left inguinal region, left anterior superior thigh, left distal thigh, proximal tibia and dorsal aspect of the right foot (Fig. 2).
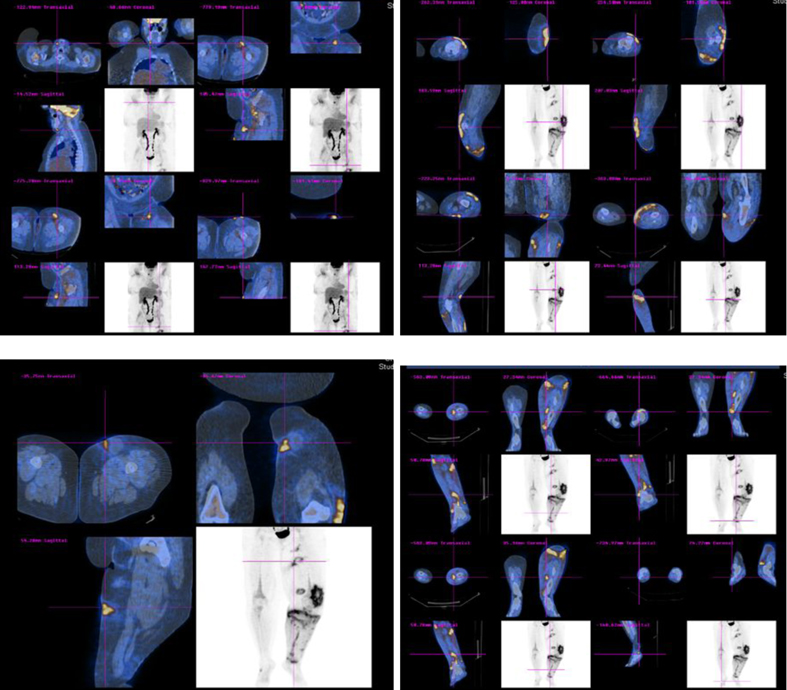 Click for large image | Figure 2. Whole body PET-CT scan showing multiple areas of hypermetabolic activity. |
The patient was started on oral bexarotene (a retinoid X receptor activator) once daily (300 mg/m2). Although the patient tolerated bexarotene well, she developed medication-induced hypothyroidism (TSH 19 µIU/mL). A repeat whole body PET-CT scan three months later showed progression of her lymphoma (Fig. 3). However, a repeat bone marrow biopsy revealed no evidence of marrow involvement by lymphoma.
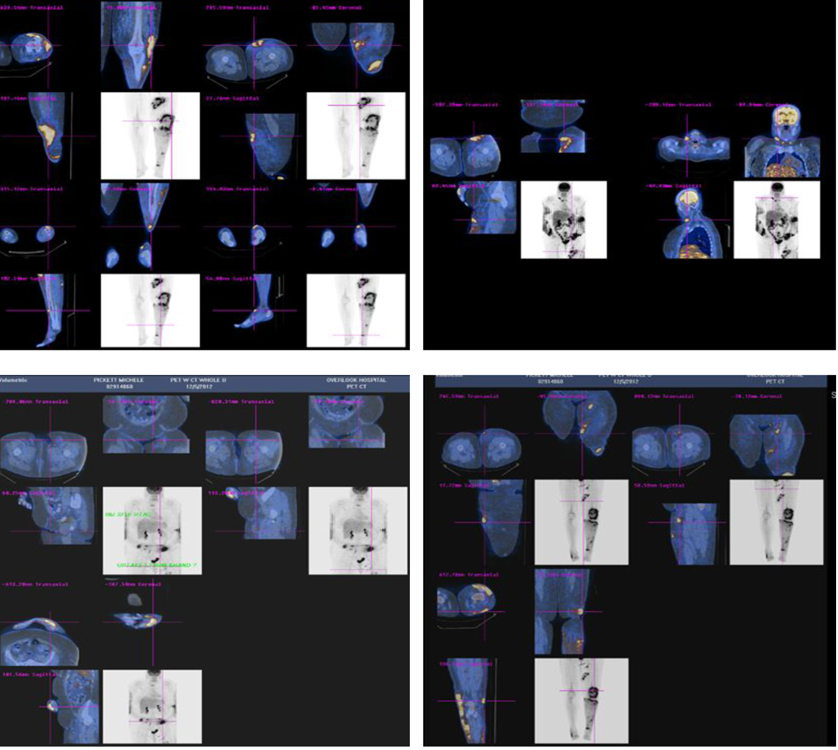 Click for large image | Figure 3. Whole body PET-CT scan showing worsening of lymphoma with increase hypermetabolic activity in lower extremities, submandibular region and abdomen. |
Because of progression of her lymphoma while on oral bexarotene, the patient was started on romidepsin. The adjusted dose of romidepsin for her body surface area (BSA) was 25 mg intravenously (14 mg/m2) on days 1, 8 and 15 of a 28-day cycle.
Following the first dose of her first cycle, the patient complained of some nausea and vomiting. Serum creatinine increased from 1.1 mg/dL (baseline) to 2.5 mg/dL, while her urinalysis and 24-hr urinary protein were within normal limits (Fig. 4). Her abnormal kidney function was thought to be secondary to dehydration and she was treated with intravenous fluids. Following hydration, her serum creatinine returned to 1.0 mg/dL (baseline) over the next four days.
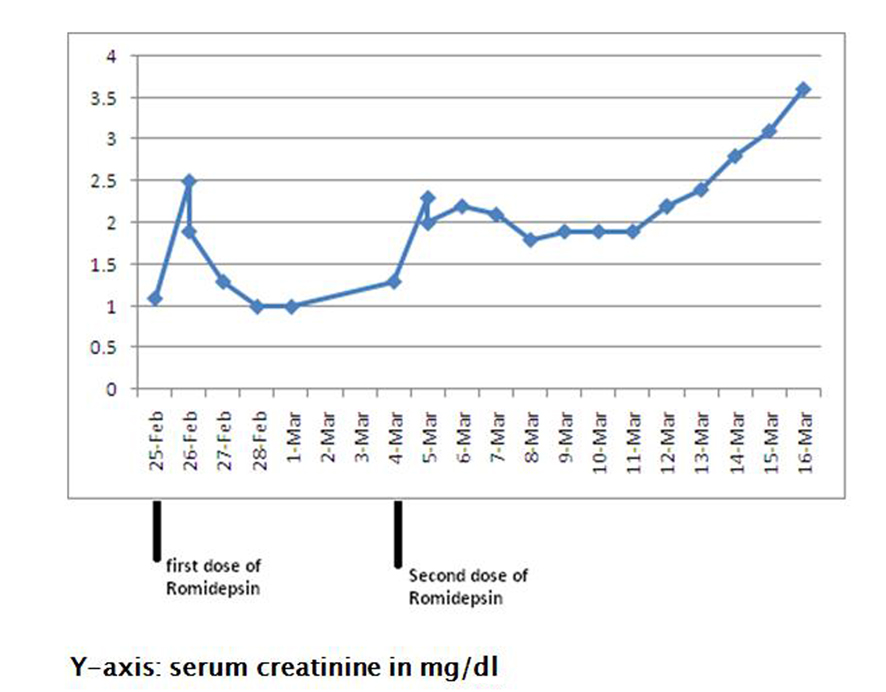 Click for large image | Figure 4. Showing rise in serum creatinine from 1.1 mg/dL (baseline) to 3.6 mg/dL following romidepsin therapy. |
One week later, the patient received her second dose (first cycle) of romidepsin. Since she had complained of nausea and vomiting following the first dose, her second dose was decreased to 19 mg intravenously (10 mg/m2). The following day, she complained of fever, chills, nausea and vomiting. On examination, worsening bilateral lower extremity edema was noted. Urinalysis showed large amounts of protein (> 1,000 mg/dL), and her 24-hr urine showed nephrotic-range proteinuria with a protein excretion of 12.6 g/day. Serum albumin ranged from 1.2 - 1.8 g/dL, and serum creatinine continued to increase from 1.1 mg/dL (baseline) to 3.6 mg/dL over a period of twelve days (Fig. 4). Anti-nuclear antibody, complement levels and uric acid were within normal limits. Romidepsin was stopped, and a nephrologic consultation was obtained.
Renal biopsy was performed, and results revealed focal collapsing glomerulopathy, diffuse severe acute tubular injury, diffuse interstitial inflammation and interstitial edema, and mild to moderate arteriosclerosis (Fig. 5).
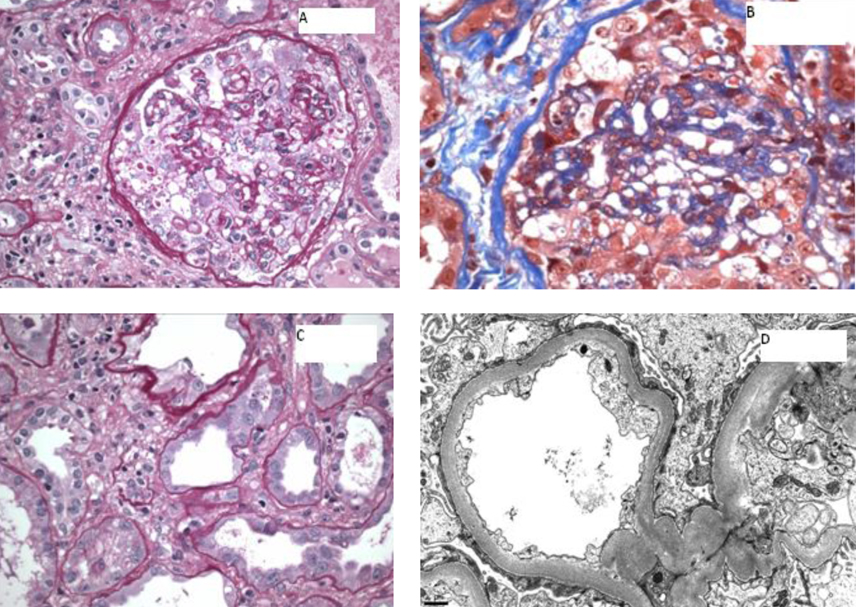 Click for large image | Figure 5. A) A glomerulus showing global implosive collapse of capillary lumina and prominent hyperplasia of epithelial cells, some of which contain prominent protein droplets (PAS stain; original magnification х 200); B) A glomerulus showing implosive collapse lumina and prominent hyperplasia of epithelial cells (Masson’s trichrome stain; original magnification х 400); C) Proximal tubules displaying loss of apical brush border and cytoplasmic attenuation (PAS stain, original magnification х 400); D) Electron photomicrograph showing diffuse foot process effacement (10,000 х). |
The patient was treated with intravenous high dose steroids (dexamethasone 10 mg). Following treatment with steroids, her kidney function gradually started showing improvement, and her serum creatinine stabilized in the range of 1.6 - 1.8 mg/dL (Fig. 6). She was continued on a steroid taper.
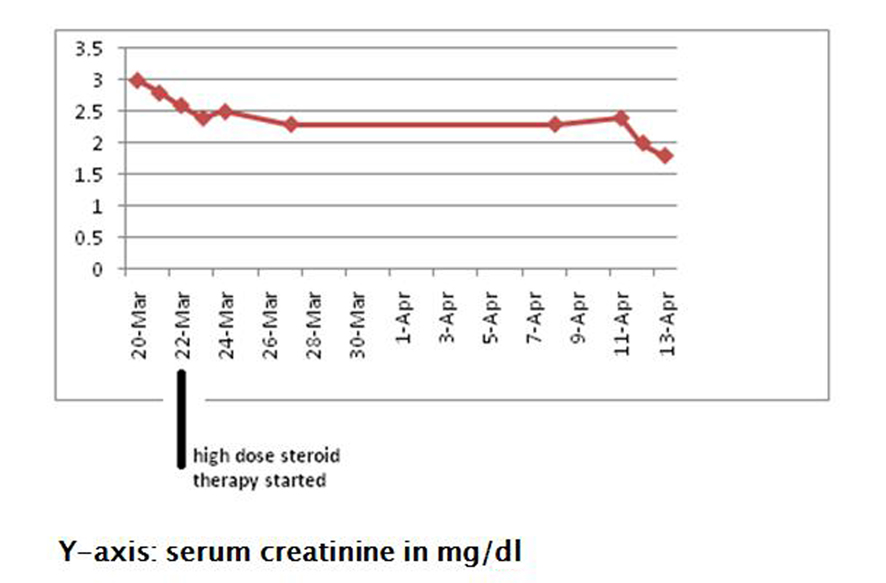 Click for large image | Figure 6. Showing improvement in serum creatinine following treatment with high dose steroids. |
Three months later, a repeat whole body PET-CT scan showed further progression of her lymphoma. Currently, the patient is being treated with panobinostat, and has shown good response to this medication.
| Discussion | ▴Top |
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare cytotoxic T-cell lymphoma seen primarily in young women [1-4]. Patients typically present with multiple subcutaneous tender nodules on the trunk and extremities. Systemic manifestations include fever, weight loss, generalized fatigue, pancytopenia, hepatitis and lymphadenopathy. The median 5-year survival is greater than 80%, except for patients with hemophagocytic syndrome (HPS) [1-3]. HPS is seen in up to 20% of presentations of SPTCL and usually carries a poor prognosis. Symptoms typically include fever, cytopenias, splenomegaly, hypertriglyceridemia, low fibrinogen levels, increased interleukin-2 receptors, hemophagocytosis and decreased activity of natural killer cells [5, 6]. Various agents have been used to treat SPTCL but because of the rarity of this lymphoma, no standard guidelines for treatment exist. Indolent local disease presentations can be treated with radiotherapy [7, 8]. Indolent local disease with more generalized presentation can be treated with retinoids such as bexarotene. Side effects include hypothyroidism and hypertriglyceridemia. Monotherapy with steroids has also been used successfully to treat SPTCL [7, 9]. However, aggressive disease presentations are treated with high dose chemotherapy followed by stem cell transplantation [7, 8, 10].
Romidepsin is a potent histone deacetylase inhibitor, a bicyclic depsipeptide isolated from the bacterium Chromobacterium violaceum. It was approved by the U.S. Food and Drug Administration (FDA) on November 5, 2009, for the treatment of cutaneous T-cell lymphoma. Although the exact mechanism of action is unknown, its enhancement of cell cycle arrest, apoptosis and alteration in tumor suppressor gene transcription is thought to play a role in its mechanism. Romidepsin is indicated for treatment in patients with cutaneous T-cell lymphoma who have failed one systemic therapy. The standard dose is 14 mg/m2 administered on days 1, 8 and 15 of a 28-day cycle. Side effects typically include nausea, vomiting, gastrointestinal disturbances, anemia, thrombocytopenia, altered taste perception, electrolyte imbalance and hypocalcemia [11, 12]. Although reversible ST/T changes and mild reversible arrhythmias have been reported with romidepsin therapy, no significant changes in left ventricular ejection fraction were seen [13]. Reversible ageusia secondary to romidepsin has also been reported [14]. However, glomerular injury secondary to romidepsin has not been reported to date.
Focal segmental glomerulosclerosis (FSGS) is a histologic lesion expressed in a variety of primary and secondary glomerular diseases, and there has been an increase in the frequency of its diagnosis worldwide over the past few decades. It is most commonly seen in the United States in those of African descent [15, 16].
This lesion has been further categorized into cellular, collapsing, peri-hilar, tip, and classic (not otherwise specified) types, with collapsing FSGS usually associated with severe nephrotic syndrome, a worse prognosis, and more resistance to treatment [17]. Although collapsing FSGS is most often associated with HIV infection, the incidence of non-HIV associated FSGS has increased in recent years. Most cases of collapsing FSGS not associated with HIV infection are idiopathic but other infections, drugs, toxins, autoimmune diseases, and malignancies have also been associated with collapsing FSGS. Injury to the podocytes and parietal epithelial cells with the subsequent loss of the glomerular filtration barrier function represents the common underlying mechanism.
A number of familial forms of FSGS have been described [18-20]. These account for a significant proportion of patients with steroid-resistant disease [19]. Collapsing FSGS has been described in a number of infections other than HIV, such as parvovirus B19, pulmonary tuberculosis, cytomegalovirus infection, and leishmaniasis. Collapsing FSGS has also been reported in patients with lupus. In a retrospective study by Salvatore et al, kidney biopsies performed in patients with lupus (over a period of seven years) revealed 19 HIV-negative patients with collapsing FSGS [21]. All patients had active lupus flares at the time of biopsy and more than half the patients developed end stage renal disease within two years. Although the exact mechanism remains unknown, an association with polymorphism in the APOL1 gene seems to play a role. Rare cases of collapsing FSGS have also been reported with sickle cell anemia and adult-onset Still’s disease [22, 23]. Histopathological and clinical features of collapsing FSGS have been described in patients with IgA nephropathy [24].
Collapsing FSGS has been reported in patients with multiple myeloma and breast cancer after treatment with higher than usual recommended doses of the bisphosphonate pamidronate and has since been described with other medications in this class [25]. Drugs such as heroin and lithium are also known to cause FSGS [26, 27]. Severe obesity, preeclampsia and long term use of anabolic steroids has been associated with FSGS [28-30]. Interferon (alpha, beta and gamma) has also been associated with FSGS [31]. De novo cases of collapsing FSGS have been reported in patients with renal transplantation [32]. FSGS NOS and collapsing variant have been reported with anthracycline therapy [33].
Paraneoplastic glomerulonephritis is a rare complication of malignancy. Solid tumor-associated membranous nephropathy and Hodgkin’s lymphoma-associated minimal change disease are “classic” examples of paraneoplastic glomerulonephritides. Focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis, IgA nephropathy and rapidly progressive glomerulonephritis have also been associated with various malignancies. Paraneoplastic glomerulonephritides are more commonly associated with chronic lymphoid malignancies than with acute lymphocytic leukemia [34]. In addition to being associated with Hodgkin’s lymphoma, non-Hodgkin’s lymphoma and chronic lymphocytic leukemia, specific types of glomerulonephritis have been reported with cutaneous T-cell lymphoma and hairy cell leukemia [35]. Hematological diseases associated with collapsing FSGS include acute monoblastic leukemia, multiple myeloma and natural killer cell leukemia [36, 37]. Paraneoplastic FSGS in lung adenocarcinoma has been reported [38]. Furthermore, collapsing FSGS can be associated with the hemophagocytosis syndrome, an activation of bone marrow histocytes which phagocytose blood cell precursors and can be seen with underlying infections, autoimmune diseases, and lymphomas.
Minimal change disease (MCD) is the most common glomerulopathy associated with Hodgkin’s lymphoma. It occurs in less than 1% of patients with Hodgkin’s lymphoma. Focal segmental glomerulosclerosis has also been associated with Hodgkin’s lymphoma and its occurrence is rarer than minimal change disease. In a retrospective study by Audard et al, both Hodgkin’s lymphoma and MCD responded well to chemotherapy [39]. Similarly, an excellent response to chemotherapy has been observed in patients with Hodgkin’s lymphoma-associated FSGS [34]. Membranoproliferative glomerulonephritis and membranous nephropathy are more commonly associated with chronic lymphocytic leukemia, hairy cell leukemia and non-Hodgkin’s lymphoma. IgA nephropathy has only been reported with cutaneous T-cell lymphoma [35].
Typically, paraneoplastic glomerular disease occurs in the presence of a malignancy, shows improvement with chemotherapy, and tends to recur with recurrence of malignancy. Unlike idiopathic glomerular disease, paraneoplastic glomerulonephritides are usually resistant to treatment with steroids and immunosuppressant drugs. Resolution of FSGS in a chronic lymphocytic leukemia patient following treatment with fludarabine, cyclophosphamide and rituximab has been reported in literature [40].
Despite the association between paraneoplastic glomerulonephritides and lymphoma, we strongly believe that our patient developed FSGS secondary to romidepsin therapy, for the following reasons: firstly, our patient developed FSGS immediately following the second dose of romidepsin; and secondly, unlike paraneoplastic glomerulonephritides, FSGS in our patient responded well to steroids, even though the lymphoma continued to worsen on chemotherapy.
Our case highlights collapsing variant FSGS (collapsing glomerulopathy) as a potential side effect of romidepsin therapy. We believe this is the first report of glomerular disease (FSGS) secondary to romidepsin therapy.
Conclusion
Romidepsin is used for treating patients with cutaneous T-cell lymphoma who have failed treatment with at least one systemic drug. Focal segmental glomerulosclerosis secondary to romidepsin therapy has not been reported to date. To our knowledge, this is the first reported case of collapsing FSGS following romidepsin therapy.
Conflict of Interests
The authors declare that they have no conflict of interests.
| References | ▴Top |
- Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C, Canninga-van Dijk MR, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111(2):838-845.
doi pubmed - Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. (IARC Press, Lyon, France), pp 294-295.
- Cerroni L, Kerl H, Gatter K. An illustrated guide to skin lymphoma. 2nd ed oxford (UK): Blackwell science; 2004.
- Kumar S, Krenacs L, Medeiros J, Elenitoba-Johnson KS, Greiner TC, Sorbara L, Kingma DW, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397-403.
doi - Koh MJ, Sadarangani SP, Chan YC, Chan MY, Tan AM, Tan SH, Tay YK, et al. Aggressive subcutaneous panniculitis-like T-cell lymphoma with hemophagocytosis in two children (subcutaneous panniculitis-like T-cell lymphoma). J Am Acad Dermatol. 2009;61(5):875-881.
doi pubmed - Gonzalez CL, Medeiros LJ, Braziel RM, Jaffe ES. T-cell lymphoma involving subcutaneous tissue. A clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15(1):17-27.
doi pubmed - Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101(6):1404-1413.
doi pubmed - Papenfuss JS, Aoun P, Bierman PJ, Armitage JO. Subcutaneous panniculitis-like T-cell lymphoma: presentation of 2 cases and observations. Clin Lymphoma. 2002;3(3):175-180.
doi pubmed - von den Driesch P, Staib G, Simon M, Jr., Sterry W. Subcutaneous T-cell lymphoma. J Am Acad Dermatol. 1997;36(2 Pt 2):285-289.
doi - Mukai HY, Okoshi Y, Shimizu S, Katsura Y, Takei N, Hasegawa Y, Kojima H, et al. Successful treatment of a patient with subcutaneous panniculitis-like T-cell lymphoma with high-dose chemotherapy and total body irradiation. Eur J Haematol. 2003;70(6):413-416.
doi pubmed - Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410-5417.
doi pubmed - Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485-4491.
doi pubmed - Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8(3):718-728.
pubmed - Cooper MR, Brewer S, Broketa G. Reversible ageusia associated with romidepsin therapy. Am J Health Syst Pharm. 2013;70(17):1502-1505.
doi pubmed - Haas M, Meehan SM, Karrison TG, Spargo BH. Changing etiologies of unexplained adult nephrotic syndrome: a comparison of renal biopsy findings from 1976-1979 and 1995-1997. Am J Kidney Dis. 1997;30(5):621-631.
doi - Braden GL, Mulhern JG, O'Shea MH, Nash SV, Ucci AA, Jr., Germain MJ. Changing incidence of glomerular diseases in adults. Am J Kidney Dis. 2000;35(5):878-883.
doi - Korbet SM, Genchi RM, Borok RZ, Schwartz MM. The racial prevalence of glomerular lesions in nephrotic adults. Am J Kidney Dis. 1996;27(5):647-651.
doi - Mathis BJ, Kim SH, Calabrese K, Haas M, Seidman JG, Seidman CE, Pollak MR. A locus for inherited focal segmental glomerulosclerosis maps to chromosome 19q13. Kidney Int. 1998;53(2):282-286.
doi pubmed - Pollak MR. The genetic basis of FSGS and steroid-resistant nephrosis. Semin Nephrol. 2003;23(2):141-146.
doi pubmed - Rana K, Isbel N, Buzza M, Dagher H, Henning P, Kainer G, Savige J. Clinical, histopathologic, and genetic studies in nine families with focal segmental glomerulosclerosis. Am J Kidney Dis. 2003;41(6):1170-1178.
doi - Salvatore SP, Barisoni LM, Herzenberg AM, Chander PN, Nickeleit V, Seshan SV. Collapsing glomerulopathy in 19 patients with systemic lupus erythematosus or lupus-like disease. Clin J Am Soc Nephrol. 2012;7(6):914-925.
doi pubmed - Ramidi GB, Kurukumbi MK, Sealy PL. Collapsing glomerulopathy in sickle cell disease: a case report. J Med Case Rep. 2011;5:71.
doi pubmed - Kumar S, Sheaff M, Yaqoob M. Collapsing glomerulopathy in adult still's disease. Am J Kidney Dis. 2004;43(5):e4-10.
doi pubmed - Cook HT. Focal segmental glomerulosclerosis in IgA nephropathy: a result of primary podocyte injury? Kidney Int. 2011;79(6):581-583.
doi pubmed - Markowitz GS, Appel GB, Fine PL, Fenves AZ, Loon NR, Jagannath S, Kuhn JA, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol. 2001;12(6):1164-1172.
pubmed - Dubrow A, Mittman N, Ghali V, Flamenbaum W. The changing spectrum of heroin-associated nephropathy. Am J Kidney Dis. 1985;5(1):36-41.
pubmed - Sakarcan A, Thomas DB, O'Reilly KP, Richards RW. Lithium-induced nephrotic syndrome in a young pediatric patient. Pediatr Nephrol. 2002;17(4):290-292.
doi pubmed - Praga M, Hernandez E, Morales E, Campos AP, Valero MA, Martinez MA, Leon M. Clinical features and long-term outcome of obesity-associated focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2001;16(9):1790-1798.
doi pubmed - Herlitz LC, Markowitz GS, Farris AB, Schwimmer JA, Stokes MB, Kunis C, Colvin RB, et al. Development of focal segmental glomerulosclerosis after anabolic steroid abuse. J Am Soc Nephrol. 2010;21(1):163-172.
doi pubmed - Nishimoto K, Shiiki H, Nishino T, Kimura T, Sasaki Y, Yamasaki M, Morikawa H, et al. Glomerular hypertrophy in preeclamptic patients with focal segmental glomerulosclerosis. A morphometric analysis. Clin Nephrol. 1999;51(4):209-219.
pubmed - Markowitz GS, Nasr SH, Stokes MB, D'Agati VD. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5(4):607-615.
doi pubmed - Kowalewska J. Pathology of recurrent diseases in kidney allografts: membranous nephropathy and focal segmental glomerulosclerosis. Curr Opin Organ Transplant. 2013;18(3):313-318.
doi pubmed - Mohamed N, Goldstein J, Schiff J, John R. Collapsing glomerulopathy following anthracycline therapy. Am J Kidney Dis. 2013;61(5):778-781.
doi pubmed - Mallouk A, Pham PT, Pham PC. Concurrent FSGS and Hodgkin's lymphoma: case report and literature review on the link between nephrotic glomerulopathies and hematological malignancies. Clin Exp Nephrol. 2006;10(4):284-289.
doi pubmed - Bajel A, Yin Lin M, Hill PA, Goodman D, McCormack C, Foley P, Davison J, et al. IgA nephropathy associated with cutaneous T cell lymphoma. Leuk Lymphoma. 2009;50(12):2083-2085.
doi pubmed - Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. 1999;56(6):2203-2213.
doi pubmed - Palma Diaz MF, Pichler RH, Nicosia RF, Alpers CE, Smith KD. Collapsing glomerulopathy associated with natural killer cell leukemia: a case report and review of the literature. Am J Kidney Dis. 2011;58(5):855-859.
doi pubmed - Tadokoro A, Ishii T, Takahama T, Watanabe N, Takano K, Kanaji N, Imataki O, et al. Paraneoplastic focal segmental glomerulosclerosis in a patient with lung adenocarcinoma. Intern Med. 2013;52(17):1953-1956.
pubmed - Audard V, Larousserie F, Grimbert P, Abtahi M, Sotto JJ, Delmer A, Boue F, et al. Minimal change nephrotic syndrome and classical Hodgkin's lymphoma: report of 21 cases and review of the literature. Kidney Int. 2006;69(12):2251-2260.
doi pubmed - Arampatzis S, Giannakoulas N, Liakopoulos V, Eleftheriadis T, Kourti P, Karasavvidou F, Matsouka P, et al. Simultaneous clinical resolution of focal segmental glomerulosclerosis associated with chronic lymphocytic leukaemia treated with fludarabine, cyclophosphamide and rituximab. BMC Nephrol. 2011;12:33.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Nephrology and Urology is published by Elmer Press Inc.