

| World Journal of Nephrology and Urology, ISSN 1927-1239 print, 1927-1247 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Nephrol Urol and Elmer Press Inc |
| Journal website http://www.wjnu.org |
Original Article
Volume 9, Number 1, March 2020, pages 15-24
Characterization of Novel Truncated Variants Identified From Indian Autosomal Dominant Polycystic Kidney Disease Cases
Sonam Raja, Rana Gopal Singhb, Parimal Dasa, c
aCentre for Genetic Disorders, Institute of Science, Banaras Hindu University, Varanasi, India
bInstitute of Medical Sciences, Banaras Hindu University, Varanasi, India
cCorresponding Author: Parimal Das, Centre for Genetic Disorders, Institute of Science, Banaras Hindu University, Varanasi, India
Manuscript submitted July 6, 2019, accepted January 14, 2020
Short title: Characterization of Novel Truncated Mutation
doi: https://doi.org/10.14740/wjnu392
| Abstract | ▴Top |
Background: Autosomal dominant polycystic kidney disease (ADPKD) is the most common, late-onset and genetically heterogenous disorder. Altered genetic background is the prime cause of ADPKD. Most of the cases showed mutations in PKD1, PKD2, GANAB and DNAJB11. Previous mutation screening studies reported that PKD1 and PKD2 are the major contributors to the disease. Among all types of DNA sequence variants, majority of the variants are protein truncating in nature. Loss of normal polycystin 1 (PC1) and polycystin 2 (PC2) proteins, encoded by PKD1 and PKD2 respectively, leads to the disease manifestation.
Methods: Three cases and 100 controls were selected for mutational screening of PKD1 and PKD2 gene using Sanger sequencing. To characterize the variants, in silico (mutation taster, Align-Grantham Variation Grantham Deviation (Align-GVGD), Polymorphism Phenotyping v2 (PolyPhen2), Protein Variation Effect Analyzer (PROVEAN), Sorting Intolerant From Tolerant (SIFT), Polycystic Kidney Disease Mutation Database (PKDB), ProtScale and protein Basic Local Alignment Search Tool (BLAST-p)) as well as in vitro (Western blot) studies were performed.
Results: We have identified a total of 14 variants (three truncating, three nonsynonymous, seven synonymous and one intronic). Three deletions (PKD1: c.445_445delC, PKD2: c.854_854delG and PKD2: c.1050_1050delC) were confirmed as rare, private and novel mutations in Indian ADPKD cases. Rest 11 variants were previously reported and likely neutral in nature. Loss of conservation of amino acid, decreased hydrophobicity and decreased coil forming ability of truncated proteins were supposed to affect the protein functions. Immunoblotting verified the expression of truncated proteins in cell.
Conclusions: We can conclude that identified deletion variants were truncating in nature and hence they were pathogenic mutations responsible for disease manifestation.
Keywords: ADPKD; PKD1; PKD2; Mutation; Truncated protein
| Introduction | ▴Top |
Autosomal dominant polycystic kidney disease (ADPKD) is a late-onset and most common genetically heterogenous disorder with the worldwide prevalence of one in 500 - 1,000 births [1]. It is characterized by presence of multiple cysts in both kidneys, leading to the disruption of precise architecture of the kidney. Deformed structure of the kidneys affects the renal filtration system and hence affected individuals have abnormal level (mostly higher) of serum creatine and urea. ADPKD is the fourth most common cause of end-stage renal disease eventually leading to the dialysis and kidney transplantation [2]. Most of the families showed mutation in PKD1 and PKD2. The major contributor of ADPKD is PKD1 gene encoded 4,303 amino acid (aa) long, 460kDa glycosylated integral membrane protein polycystin 1 (PC1). The second major contributor is PKD2 gene encoded 968aa long, 110kDa channel protein polycystin 2 (PC2) [3-5]. Recently two new genes GANAB and DNAJB11 were identified in a few ADPKD cases [6, 7]. Till date, ADPKD mutation database (http://pkdb.mayo.edu) has listed 2,331 in PKD1 and 305 in PKD2 DNA sequence variants [8]. PC1 and PC2 are involved in several cellular events like cell proliferation, differentiation, apoptosis, cell polarity, adhesion, maturation, tubulogenesis and extracellular matrix maintenance, which are all essential steps of kidney morphogenesis [9, 10]. Experiments on pkd1-/-null mice indicated that PC1 played an important role in renal tubule maturation [11]. Overlapping of sub-cellular localization of PC1 and PC2 suggests their mutual function in different signaling pathways [12]. It has been reported that PC1-PC1 homomer interacted via PKD (Ig like) domains, PC2-PC2 interacted via N-terminal region and PC1-PC2 heteromeric interaction occurred via C-terminal coiled-coil domains [13]. Previous reports pointed out that PKD1 and PKD2 harbored high number of protein truncating mutations [14-21]. That is why both genetic testing as well as in vitro studies are very important to confirm and verify the cause of ADPKD. Here, we reported and confirmed novel truncating variants identified in three different Indian ADPKD cases. These variants were expected to encode the smallest truncated protein. One was discovered from this study in PKD1 (c.445_445delC) gene and two were reported in PKD2 (c.854_854delG and c.1050_1050delC) gene from previous study [22]. These variants were expected to encode the smallest truncated protein which might be one of the causes of disease condition.
| Materials and Methods | ▴Top |
Patient recruitment and mutation detection
This study was approved by Institutional Ethical Committee of the Institute of Science, Banaras Hindu University, Varanasi (Institution Ethical Review board approval number: F.Sc./Ethics Committee/2015-16/6). Three clinically suspected individuals with bilateral polycystic kidney disease and 100 control individuals participated in this study with informed consent. Genomic DNA was extracted from peripheral blood samples by salting out method. The quantity and quality of extracted DNA were assayed by spectrophotometer (Nanodrop 2000) as well as 0.8% agarose gel electrophoresis. PKD1 and PKD2 gene specific primers [5, 17, 18] were used for screening of three patients. PKD2 gene screening of two cases was previously performed and reported [21]. Long range-polymerase chain reaction (LR-PCR) followed by second generation nested PCR (nPCR) was performed for PKD1 gene screening. LR-PCR product was run on 1% agarose gel and DNA was extracted according to prescribed manufacture protocol using RBC DNA elution kit (Real Genomics). Eluted DNA products were diluted in 1:1,000 deionized water and then used as a template to perform nPCR following previous protocol [5, 17, 18]. PCR products were cleaned by using Exonuclease I and rShrimp alkaline phosphatase enzyme (USB Affimetrix, Santa Clara, CA, USA). Sequencing of PCR product with forward and/or reverse primer was achieved following ABI Big Dye Terminator V3.1 cycle sequencing kit (Applied Biosystem, Foster City, CA, USA). Sequencing results were compared with reference genomic sequence NC_000016.10, mRNA sequence NM_001009944.2 and protein sequence NP_001009944.2 for PKD1 and NC_000004.12, mRNA sequence NM_000297.4 and protein sequence NP_000288.1 for PKD2 available on NCBI using basic local alignment search tool (BLAST). Discovered pathogenic variants were also screened in 100 controls.
In silico analysis
In silico analysis was performed using online software like mutation taster, Align-Grantham Variation Grantham Deviation (Align-GVGD), Polymorphism Phenotyping v2 (PolyPhen2), Protein Variation Effect Analyzer (PROVEAN), and Sorting Intolerant From Tolerant (SIFT), and was also compared with ADPKD mutation database (Table 1). Amino acid conservation of novel PC1 and PC2 variants was analyzed using protein BLAST (BLAST-p). Hydrophobicity of wild and mutant proteins was compared by ProtScale (Hopp & Woods). Secondary structure forming capability of wild and mutant proteins was analyzed using ProtScale (Chou & Fasman).
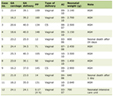 Click to view | Table 1. List of Variants Identified in Three Patients |
Plasmid preparation
Two plasmids with PKD1 and PKD2 genes were procured from Addgene (https://www.addgene.org) for in vitro studies. Due to difficulty in site-directed mutagenesis (SDM) and transfection of big-size wild-type green fluorescence protein (GFP-WT) (AF-41) plasmid, sub-cloning of 2kb region of PKD1 in pcDNA3.1 plasmid was performed. GFP-WT (AF-41) was used as a template to amplify wild-type 2kb PKD1 gene fragment which covered exon1-5, tagged with GFP at N-terminal. Sub-cloning of amplified 2kb fragment into pcDNA3.1 mammalian expression vector was completed using forward primer with BamHI and reverse primer with XhoI cut site (Table 2). This plasmid worked as a template in SDM experiment. SDM primers were designed using NEB mutagenesis primer designing web server (Table 2). SDM primer introduced the deletion of G nucleotide during the SDM-PCR using Q5 site directed mutagenesis kit (NEB, Ipswich, MA, USA). The SDM-PCR was performed in ABI thermal cycler (ABI, Foster City, CA, USA), programmed with initial denaturation at 98 °C for 2 min, followed by 35 cycles of denaturation at 98 °C for 30 s and annealing-extension at 72 °C for 2 min and one final extension of 72 °C for 5 min. NEB10 beta (NEB10B) cells were transformed with wild and mutant plasmids. Positive colonies were picked, grown in LB+ ampicillin culture and then plasmids were extracted using Qiagen plasmid extraction kit. SDM created mutant plasmid DNA (pcDNA 3.1+2kb ex4delC PKD1) was confirmed by digestion, by colony PCR amplification of the insert and Sanger sequencing. Similarly, two variants from PKD2 (c.854_854delG and c.1050_1050delC) were selected for SDM. M-PKD2 (OF2-3) plasmid having wild-type PKD2 was used as a template to introduce the deletion using SDM primers and Q5 site directed mutagenesis kit (Table 2). NEB10B competent cells were transformed with the wild M-PKD2 (OF2-3) plasmid as well as SDM made mutant plasmids (pcDNA 3.1+2kb ex4delC PKD1, M-PKD2-c.854delG and M-PKD2-c.1050delC). Positive clones were confirmed by PCR amplification of the PKD2 insert and by Sanger sequencing.
Click to view | Table 2. List of Primers Used in Sub-Cloning and SDM |
Transfection of COS7 cells, protein isolation and Western blotting
Transfection was carried in 6-wells plate seeded with 3 × 105 COS7 cells/well. At around 90% of confluency, Dulbecco’s minimum essential medium (DMEM) was replaced with the Opti-MEM (Invirogen, Thermo Fisher Scientific, Waltham, MA, USA) and then cells were transfected with the wild and mutant plasmids using Lipofectamine 2000 (Invirogen, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer protocol. For PKD1 gene variant study, COS7 cells were transfected with the wild (pcDNA 3.1+2kb wild PKD1) and mutant (pcDNA 3.1+2kb ex4delG PKD1) plasmids of PKD1 gene. Similarly, for PKD2 gene variant study, COS7 cells were transfected with the wild (M-PKD2 (OF2-3)) and mutant (M-PKD2-c.854delG and M-PKD2-c.1050delC) plasmids. After 40 h of transfection, total cellular protein was isolated and protein concentration was estimated by using Bradford reagent (Sigma-Aldrich, St. Louis, MO, USA) following the manufacturer protocol. A total of 10% SDS-PAGE was used to resolve the proteins. A total of 50 µg of denatured protein was loaded into each well and electrophoresed at 100 volts (constant) for 4 h at 4 °C. Proteins were transferred from gel to polyvinylidene fluoride (PVDF) membrane. After transfer, membrane was treated with blocking buffer (having 5% non-fat milk) at room temperature for 2 h. Blocking membrane was incubated with primary antibody at 4 °C overnight. For PC1 protein detection, membrane was incubated with 1:1,000 dilution of primary antibody anti-PC1 raised in mouse (Santa Cruz, CA, USA) and anti-GFP raised in mouse (Santa Cruz, CA, USA). For PC2 protein detection, membrane was incubated with 1:1,000 dilution of primary antibody anti-Myc raised in mouse (Sigma-Aldrich, St. Louis, MO, USA). The 1:1000 dilution of anti-beta actin raised in mouse (Sigma-Aldrich, St. Louis, MO, USA) primary antibody was used to detect internal loading control beta actin protein. After incubation, membrane was washed in Tris-buffered saline (TBS) three times for 10 min and incubated at room temperature for 2 h with secondary antibody. Two secondary antibodies (1:1,000 dilution), goat anti-mouse IgG-ALP (Bangalore Genei, Bangalore, India) and goat anti-mouse IgG-HRP (Bangalore Genei, Bangalore, India) were used separately to process the membrane. Membrane was washed two times with TBS for 10 min and developed by either nitro-blue-tetrazolium-5-bromo-4-chloro-3-indolyl-phosphate (NBT-BCIP) or electrochemiluminescence (ECL) technique. Protein isolated from PC1 wild and mutant plasmid transfected cells displayed bands of 72KDa and 54KDa. Protein isolated from PC2 wild and mutant plasmid transfected cells displayed bands of 110KDa, 46 KDa and 42KDa.
| Results | ▴Top |
PKD1 variant
A 42-year-old female patient (sample ID: PKD16.1) with negative family history of ADPKD had bilateral cysts for the past 9 years. She has grown an enlarged kidney of size 13 cm × 5.28 cm of the right kidney and 15.8 cm × 8.3 cm of the left kidney. Serum analysis revealed the increased level of urea (22 mg/dL) and creatinine (1.2 mg/dL). She has other complications like flank pain, loss of appetite, weakness, painful urination and urinary tract infection. Mutation screening of PKD1 and PKD2 genes revealed nine DNA sequence variants in the patient genome (Table 1). She has heterozygous c.445_445delC in PKD1 (Fig. 1) variant specifically located in N-terminal extracellular leucine rich repeat c-type lectin (LRRCT) domain of PC1. Along with the c.445_445delC pathogenic variant, there were seven in PKD1 (c.7165T>C, c.7441C>T, c.12133A>G, c.12176C>T, c.12276A>G, c.12409C>T and c.12630T>C) and one in PKD2 (IVS3-22G>A) variants discovered in patient. Absence of c.445_445delC in 100 controls and in other populations determined it as a rare and novel variant. In silico analysis revealed that c.445_445delC encoded 288aa truncated protein (p.Q149Sfs*141). Rest of the variants were not damaging in nature. Multiple sequence alignment of wild-PC1 and mutant (p.Q149Sfs*141) proteins revealed 149aa sequence similarity and after that 139aa altered sequece ultimately ending with the premature stop codon. This mutant protein was able to form the wild-type structure upto first transmembrane domain and rest of the downstream domains were either compromisezed or lost (Fig. 2). ProtScale web server suggested drop in the hydrophobic microenvironment around the mutant PC1: p.Q149Sfs*141 compared to wild-PC1 (Fig. 3a, b). Secondary structure forming capacity analyzed by ProtScale revealed decreased coil forming capacity of mutant PC1: p.Q149Sfs*141 compared to the wild-PC1 (Fig. 3c, d). It was expected that pcDNA 3.1+2kb wild PKD1 plasmid will encode the 72kDa protein and the SDM created mutant pcDNA 3.1+2kb ex4delG PKD1 plasmid will encode the 54kDa truncated protein in transfected cells. GFP and PC1 specific primary antibody detected the specific 72kDa and 54kDa protein band and verified that c.445_445delC is protein truncating in nature (Fig. 4a-f).
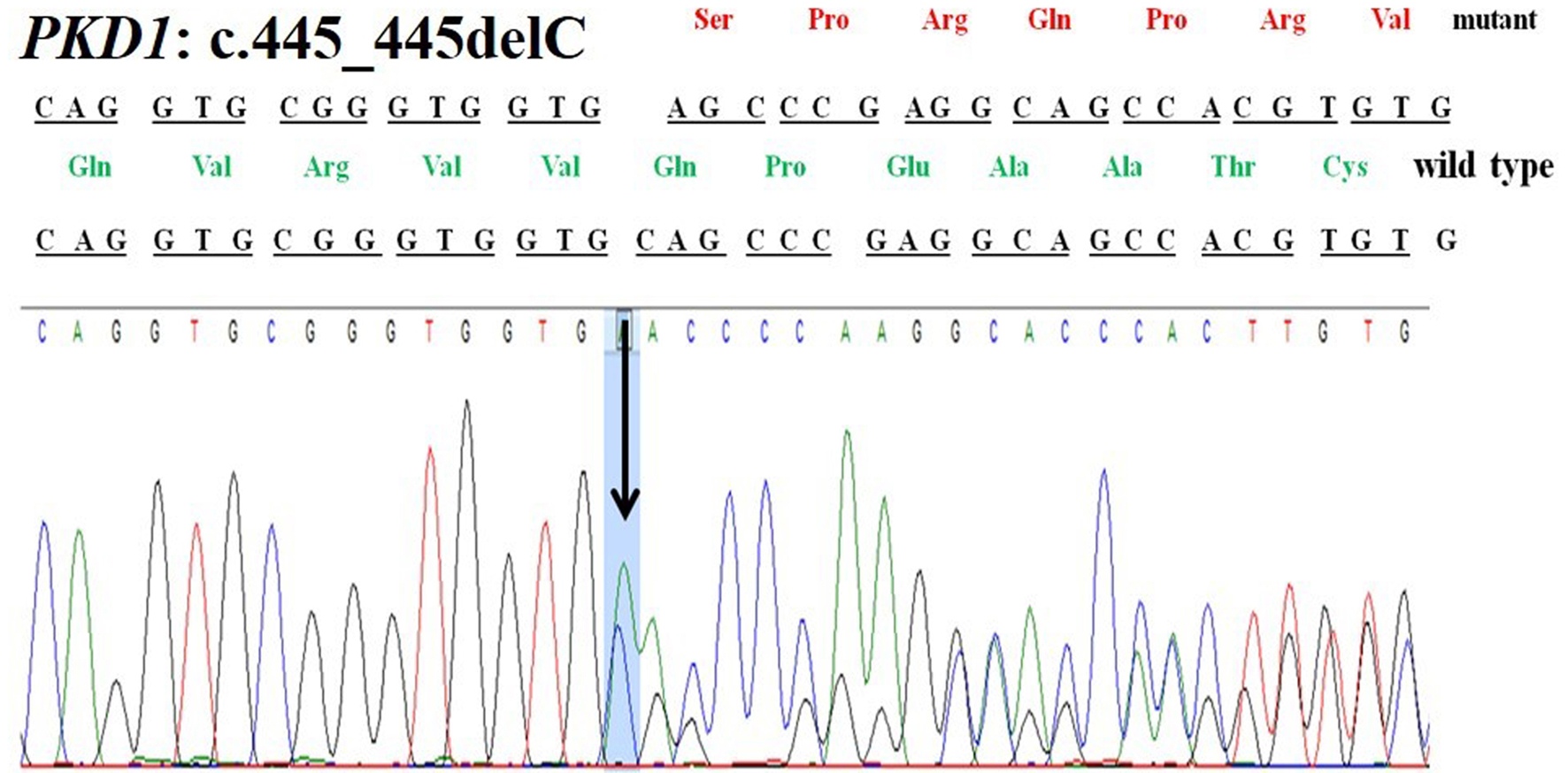 Click for large image | Figure 1. Chromatogram of PKD1: c.445_445delC deletion in patient 16.1. |
 Click for large image | Figure 2. Multiple sequence alignment of wild PC1 and predicted mutant c.445_445delC-PC1 protein. PC1: polycystin 1. |
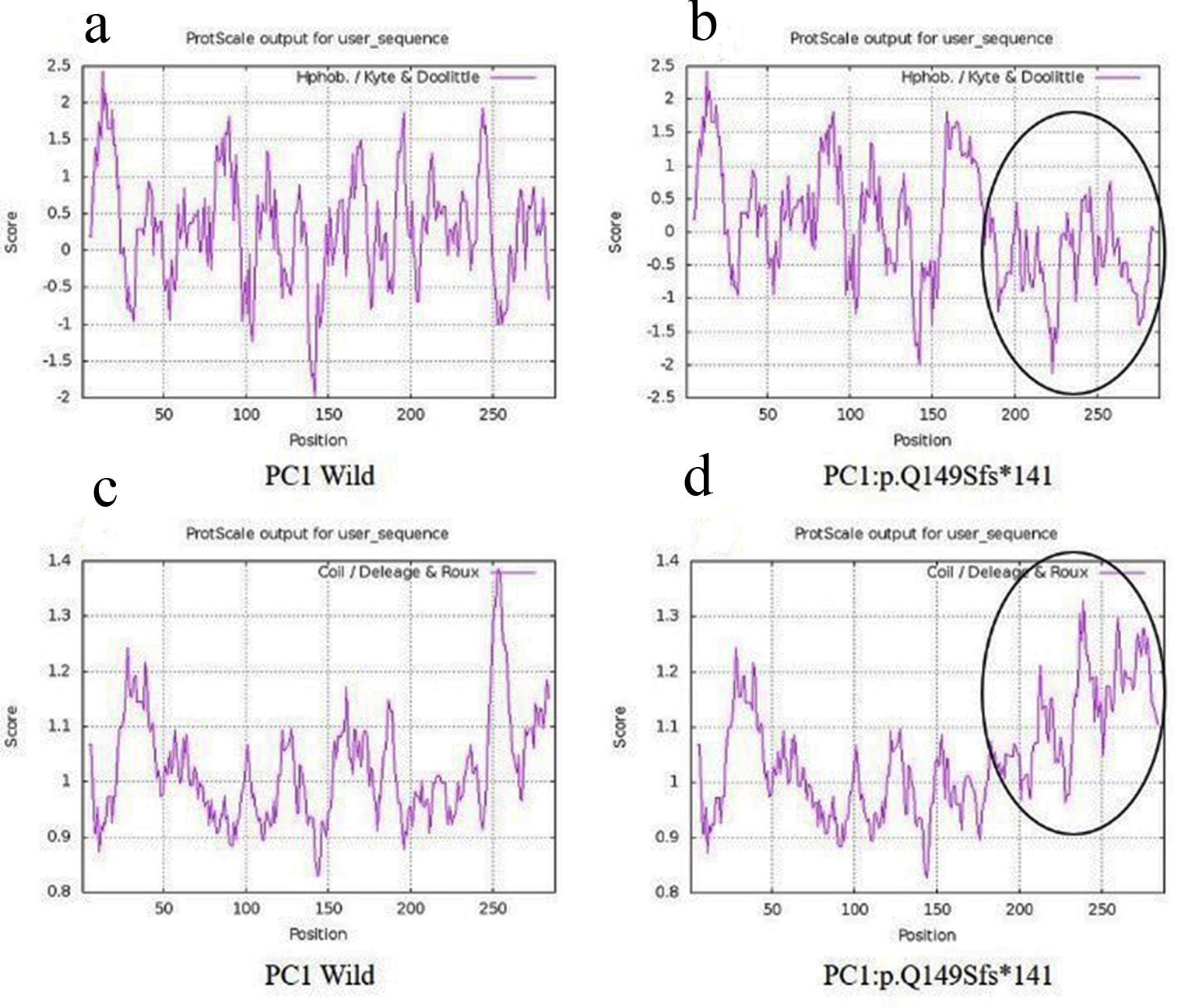 Click for large image | Figure 3. Hydrophobicity and helix forming capacity calculation of wild and mutant proteins using ProtScale. (a, b) Hydrophobicity of PC1-wild and PC1: p.Q149Sfs*141; (c, d) helix forming capacity around PC1-wild and PC1:p.Q149Sfs*141. PC1: polycystin 1. |
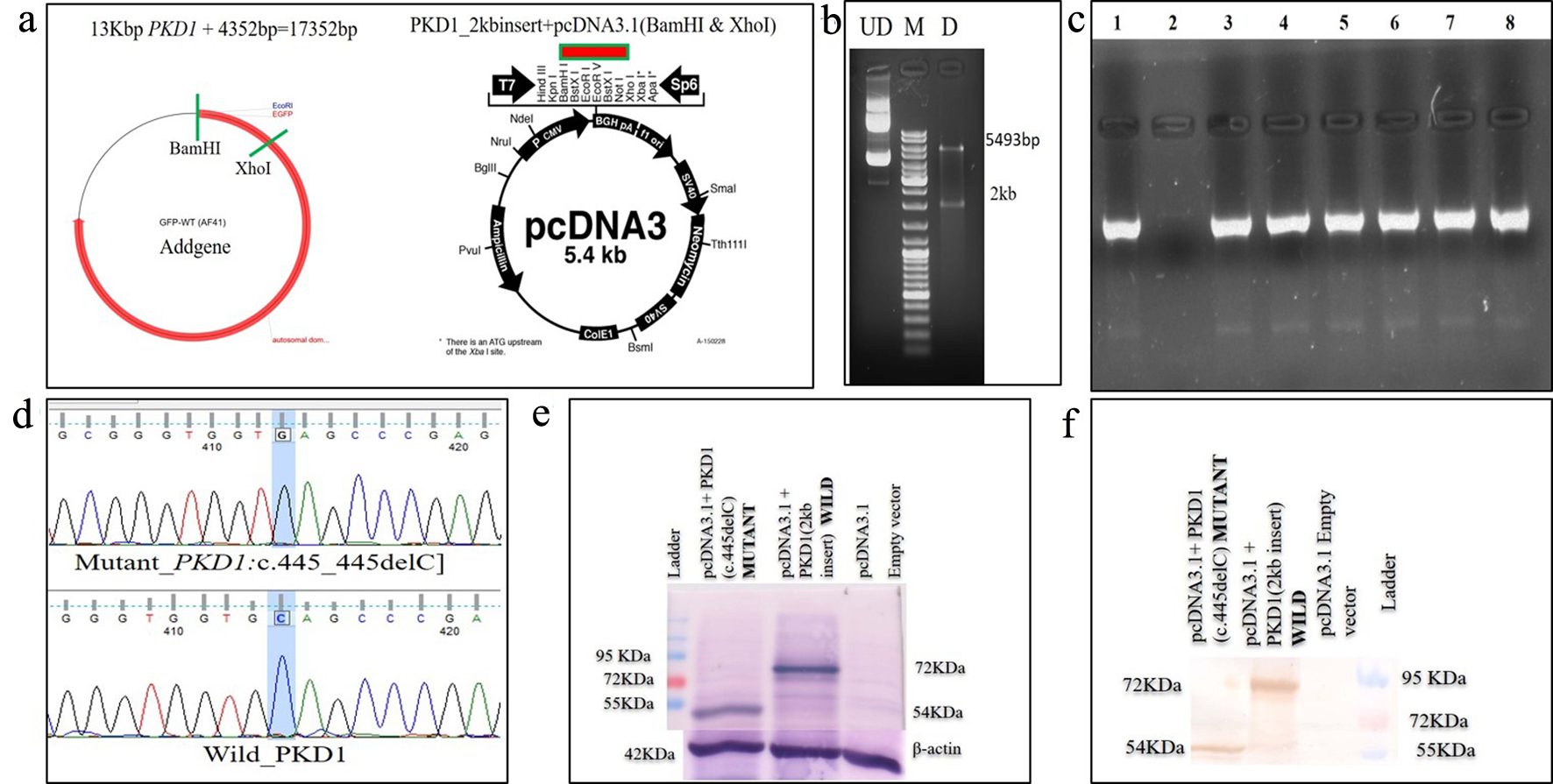 Click for large image | Figure 4. Schematic diagram of (a) excision of GFP-tagged wild PKD1 2kb region using BamHI and XhoI and sub-cloned into pcDNA3.1 mammalian expression vector; (b) confirmation of sub-cloned plasmid using restriction enzyme; (c) colony PCR to confirm SDM after transformation using PKD1 specific primer; (d) Sanger sequencing of plasmids, mutant pcDNA 3.1+2kb ex4delC PKD1, and wild pcDNA 3.1+2kb wild PKD1; (e) expression of wild and mutant proteins in transfected COS7 cells, lane 1 displaying 54kDa mutant and lane 2 displaying 72kDa mutant protein using anti-PC1 antibody, and lane 3 displaying empty vector; (f) lane 1 displayed 54kDa mutant, lane 2 displayed 72kDa mutant protein and lane 3 displayed empty vector using anti-GFP antibody. GFP: green fluorescence protein; PCR: polymerase chain reaction; SDM: site-directed mutagenesis. |
PKD2 variant
A 68-year-old female patient (sample ID: PKD39.1) with negative family history of ADPKD had complications of bilateral cysts and enlarged kidney for the past 7 years. Serum analysis showed the increased level of urea 40 mg/dL and creatinine 1.59 mg/dL. She had other clinical complications like flank pain, loss of appetite and weakness. Mutation screening revealed two variants: one in PKD1 (c.1119C>T) and another one in PKD2 (c.854_854delG) (Table 1). Heterozygous c.854_854delG variant was absent in 100 controls and other population suggested it as rare and novel. In silico analysis revealed that c.854_854delG variant was present in N-terminal first extracellular loop which was supposed to encode 317aa truncated protein p.G285Afs*32 and the c.1119C>T was likely neutral in nature.
Another 30-year-old male patient (sample ID: PKD64.1) with positive family history of ADPKD had complications of bilateral cysts for the past 4 years with enlarged kidney of size 10.8 cm × 4.6 cm of the right kidney and 14.9 cm × 7.4 cm of the left kidney. Serum analysis indicated the increased level of urea 21 mg/dL and creatinine 1.0 mg/dL. He has complications like flank pain, loss of appetite, weakness, swelling in body and hematuria. Mutation screening of PKD1 and PKD2 gene revealed three variants in the patient (Table 1). Heterozygous c.1050_1050delC variant was discovered in N-terminal first extracellular loop. Along with the c.1050_1050delC, two other PKD1 (c.1119C>T and c.10529C>T) variants were also harbored by the patient 64.1. In silico analysis revealed that c.1050_1050delC encoded 373aa truncated protein p.S351Vfs*24 and rest two variants c.1119C>T and c.10529C>T were likely neutral in nature. Multiple sequence alignment analysis of both truncated proteins (p.G285Afs*32 and p.S351Vfs*24) revealed 285aa sequence similarity with wild-PC2. The 250aa sequence was supposed to encode upto the first transmembrane domain and the remaining downstream domains were suspected to be missing in both mutant variants (Fig. 5). ProtScale web server suggested drop in the hydrophobic microenvironment around the PC2: p.G285Afs*32, PC2: p.G285Afs*32 compared to wild-PC2. Secondary structure forming capacity analyzed by ProtScale revealed decreased coil forming capacity of PC2: p.G285Afs*32, PC2: p.G285Afs*32 compared to the wild-PC2 (Fig. 6). To study the frameshift-induced protein truncation, COS7 cells were transfected with wild and mutant variant containing plasmids. It was expected that the wild PC2 will encode 110kDa PC2, c.854_854delG will encode 46kDa truncated PC2 and c.1050_1050delC will encode 42KDa truncated PC2. Primary antibody against Myc detected the 42kDa, 46kDa and 110kDa molecular weight protein band and verified both variants c.854_854delG and c.1050_1050delC as protein truncating mutations (Fig. 7a-g).
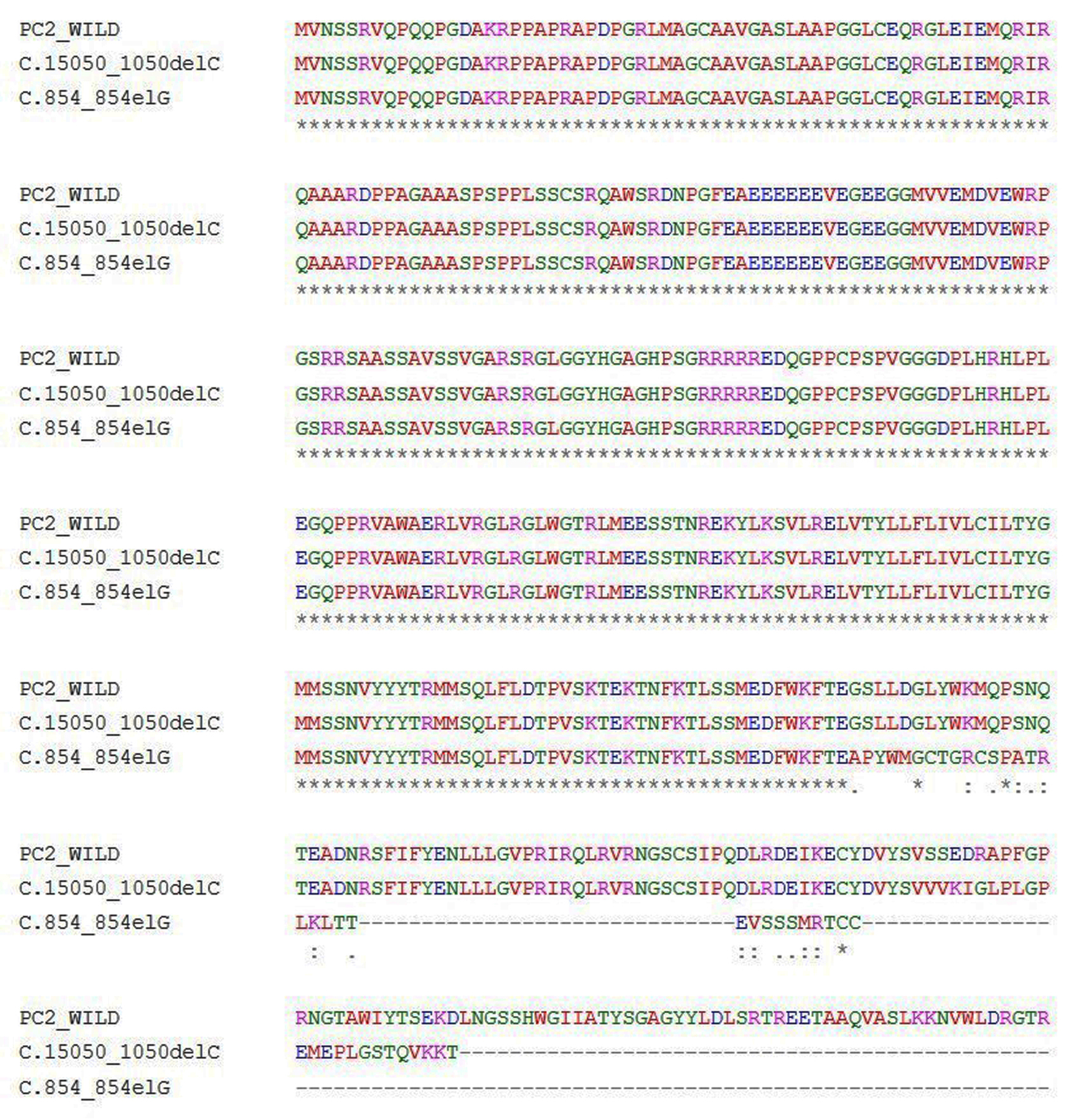 Click for large image | Figure 5. Multiple sequence alignment of wild PC2 and predicted mutants c.854_854delG-PC2 and c.1050_1050delC-PC2 proteins. |
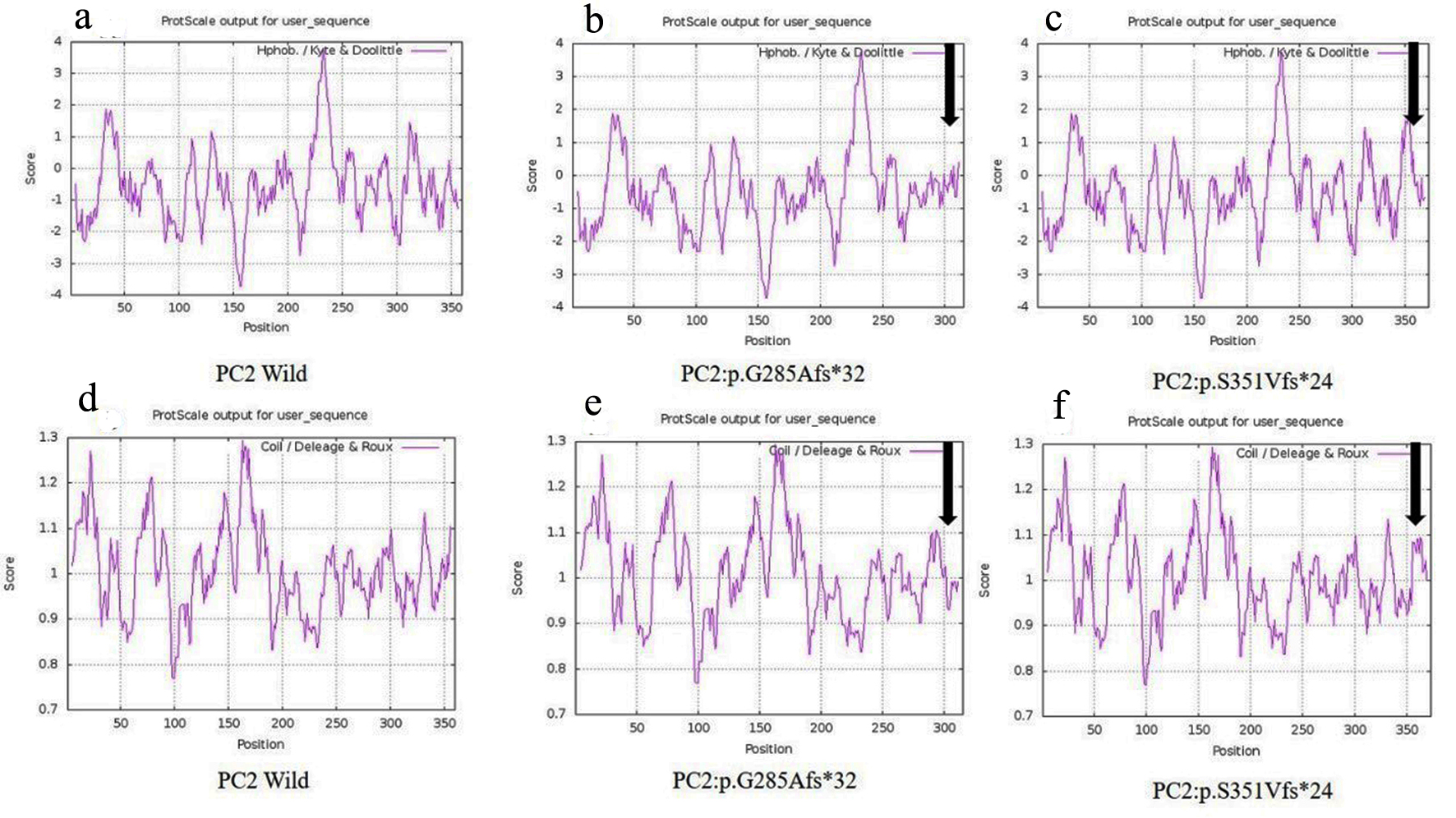 Click for large image | Figure 6. Hydrophobicity and helix forming capacity calculation of wild and mutant proteins using ProtScale. (a-c) Hydrophobicity of PC2-wild, PC2: p.G285Afs*32 and PC2: p.G285Afs*32, and (d-f) helix forming capacity of PC2-wild, PC2: p.G285Afs*32 and PC2: p.G285Afs*32. |
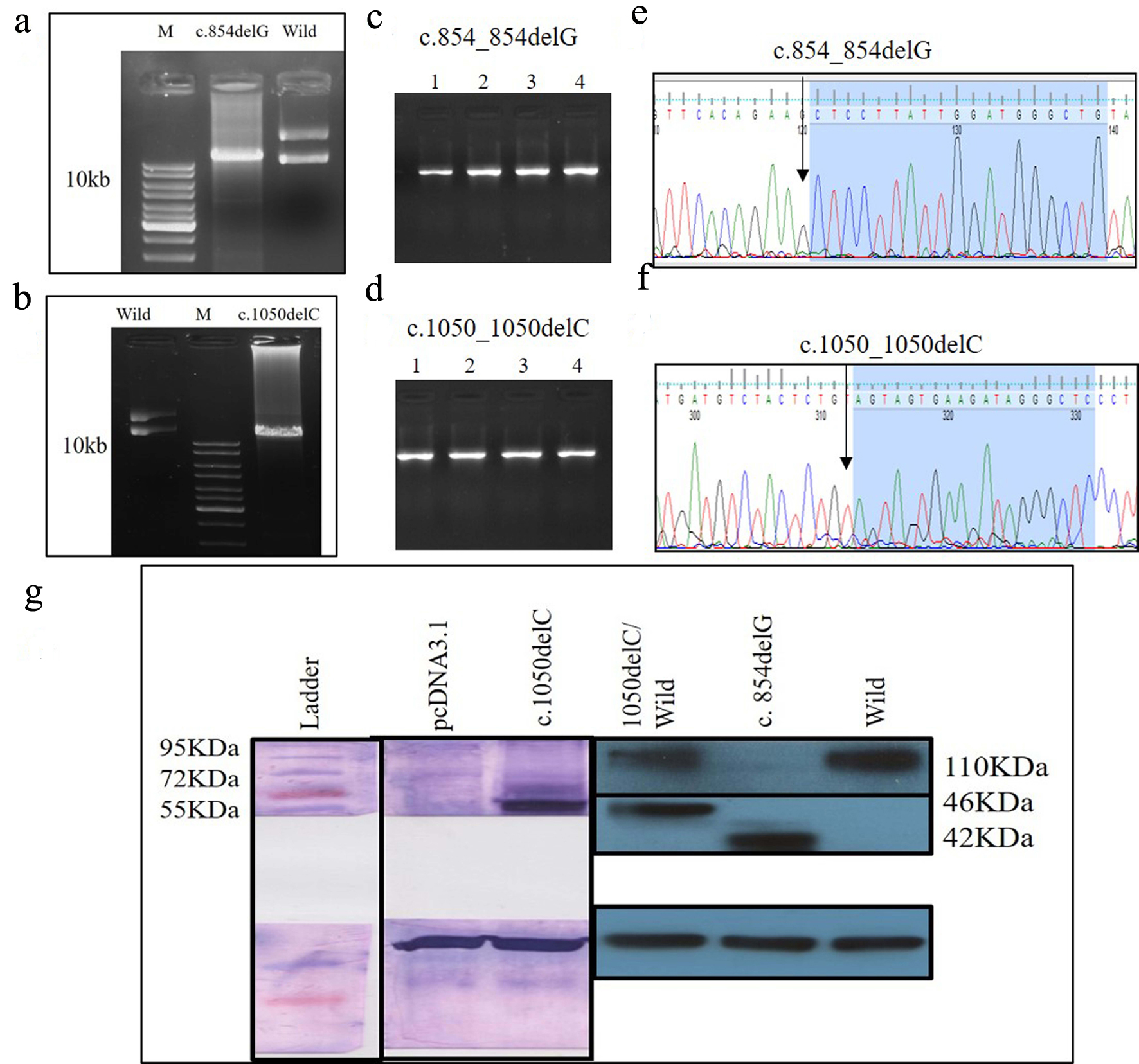 Click for large image | Figure 7. (a) PCR product of c.854_854delG SDM, (b) PCR product of c.1050_1050delC SDM, (a, d) colony PCR to confirm the transformation using PKD2 specific primer, (e, f) Sanger sequencing of plasmids mutants c.854_854delG, c.1050_1050delC, and (g) identification of wild and mutant PC2 proteins using Myc-tag antibody. PCR: polymerase chain reaction; SDM: site-directed mutagenesis; PC2: polycystin 2. |
| Discussion | ▴Top |
Previous reports published form American [15-17, 19-21], Asian [18], Indian [22] and Iranian [23] populations suggested that protein truncation is one of the prime causes of ADPKD. A total of 14 DNA sequences were identified and three deletion variants were verified to be mutation. Absence of deletion variants PKD1: c.445_445delC, PKD2: c.854_854delG and PKD2: c.1050_1050delC in controls and other populations suggested that these are rare and novel. Change in hydrophobicity and coil forming ability of truncated protein might affect the structure of the protein. In silico analysis determined all novel variants (PKD1: c.445_445delC, PKD2: c.854_854delG and PKD2: c.1050_1050delC) as damaging and rest of the 11 variants as not damaging. In silico studies revealed that PKD1 (c.445_445delC) and PKD2 (c.854_854delG and c.1050_1050delC) encoded truncated protein (p.Q149Sfs*141 of PKD1, p.G285Afs*32 and p.S351Vfs*24 of PKD2) respectively. 3D and 2D cell cultures are suitable models to study the functional impact of mutations responsible for formation and growth of cyst [24]. Immunoblotting confirmed expression of truncated mutations in vitro using 2D cell culture. The loss of one functional allele might disrupt the critical window of normal protein availability in the cell to perform the normal cellular functions like channel property and the protein-protein interaction and hence prove their pathogenicity for disease. Occurrence of pathogenic truncating variation in the patients is the cause of disease. Although there is no cure for ADPKD, specific treatment can not only ease the symptom, but can even prolong the life span. Better understanding of this disease may help to develop novel strategy for early diagnosis and treatment.
Acknowledgments
We are very thankful for participation of patients and healthy control individuals. SR is thankful to UGC, New Delhi, India for providing SRF.
Financial Disclosure
This work was supported by UGC-UPE focus area-II, Banaras Hindu University, India.
Conflict of Interest
We certify that the research findings presented here are original and free of conflict of interest.
Informed Consent
Individuals participated in this study provided informed consent.
Author Contributions
This study was designed by PD. Experiments were designed and performed by SR. Clinical diagnosis was carried out by RGS. Manuscript was drafted by SR and PD.
| References | ▴Top |
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301.
doi - Wilson PD. Polycystin: new aspects of structure, function, and regulation. J Am Soc Nephrol. 2001;12(4):834-845.
- Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The international polycystic kidney disease consortium. Cell. 1995;81(2):289-298.
doi - Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339-1342.
doi pubmed - Hayashi T, Mochizuki T, Reynolds DM, Wu G, Cai Y, Somlo S. Characterization of the exon structure of the polycystic kidney disease 2 gene (PKD2). Genomics. 1997;44(1):131-136.
doi pubmed - Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, Edwards ME, et al. Mutations in GANAB, encoding the glucosidase IIalpha subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016;98(6):1193-1207.
doi pubmed - Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol. 2018;29(1):13-23.
doi pubmed - ADPKD mutation database (http://pkdb.mayo.edu/).
- Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, Ratcliffe PJ, et al. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93(4):1524-1528.
doi pubmed - van Adelsberg JS. The role of the polycystins in kidney development. Pediatr Nephrol. 1999;13(5):454-459.
doi pubmed - Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13(24):3069-3077.
doi pubmed - Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, et al. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem. 1999;274(40):28557-28565.
doi pubmed - Newby LJ, Streets AJ, Zhao Y, Harris PC, Ward CJ, Ong AC. Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J Biol Chem. 2002;277(23):20763-20773.
doi pubmed - Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A. 1997;94(13):6965-6970.
doi pubmed - Watnick TJ, Piontek KB, Cordal TM, Weber H, Gandolph MA, Qian F, Lens XM, et al. An unusual pattern of mutation in the duplicated portion of PKD1 is revealed by use of a novel strategy for mutation detection. Hum Mol Genet. 1997;6(9):1473-1481.
doi pubmed - Watnick T, Phakdeekitcharoen B, Johnson A, Gandolph M, Wang M, Briefel G, Klinger KW, et al. Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very-early-onset disease. Am J Hum Genet. 1999;65(6):1561-1571.
doi pubmed - Phakdeekitcharoen B, Watnick TJ, Germino GG. Mutation analysis of the entire replicated portion of PKD1 using genomic DNA samples. J Am Soc Nephrol. 2001;12(5):955-963.
- Phakdeekitcharoen B, Watnick TJ, Ahn C, Whang DY, Burkhart B, Germino GG. Thirteen novel mutations of the replicated region of PKD1 in an Asian population. Kidney Int. 2000;58(4):1400-1412.
doi pubmed - Neumann HP, Bacher J, Nabulsi Z, Ortiz Bruchle N, Hoffmann MM, Schaeffner E, Nurnberger J, et al. Adult patients with sporadic polycystic kidney disease: the importance of screening for mutations in the PKD1 and PKD2 genes. Int Urol Nephrol. 2012;44(6):1753-1762.
doi pubmed - Roelfsema JH, Spruit L, Saris JJ, Chang P, Pirson Y, van Ommen GJ, Peters DJ, et al. Mutation detection in the repeated part of the PKD1 gene. Am J Hum Genet. 1997;61(5):1044-1052.
doi pubmed - Rossetti S, Strmecki L, Gamble V, Burton S, Sneddon V, Peral B, Roy S, et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet. 2001;68(1):46-63.
doi pubmed - Raj S, Singh RG, Das P. Mutational screening of PKD2 gene in the north Indian polycystic kidney disease patients revealed 28 genetic variations. J Genet. 2017;96(6):885-893.
doi pubmed - Bitarafan F, Garshasbi M. Molecular genetic analysis of polycystic kidney disease 1 and polycystic kidney disease 2 mutations in pedigrees with autosomal dominant polycystic kidney disease. J Res Med Sci. 2019;24:44.
doi pubmed - Sharma M, Reif GA, Wallace DP. In vitro cyst formation of ADPKD cells. Methods Cell Biol. 2019;153(93-111.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Nephrology and Urology is published by Elmer Press Inc.